Class I医疗器械美国上市,制造商应注意...
由于对患者而言风险最低,Class I是最快进入美国市场的医疗器械类别,大部分Class I器械不受FDA上市前通知510(k)或上市前批准PMA的要求所约束。
Class I器械受FDA的一般控制,是适用于所有医疗器械的一系列控制措施,涉及:器械注册、掺假、品牌错误、维护记录和良好制造规范。
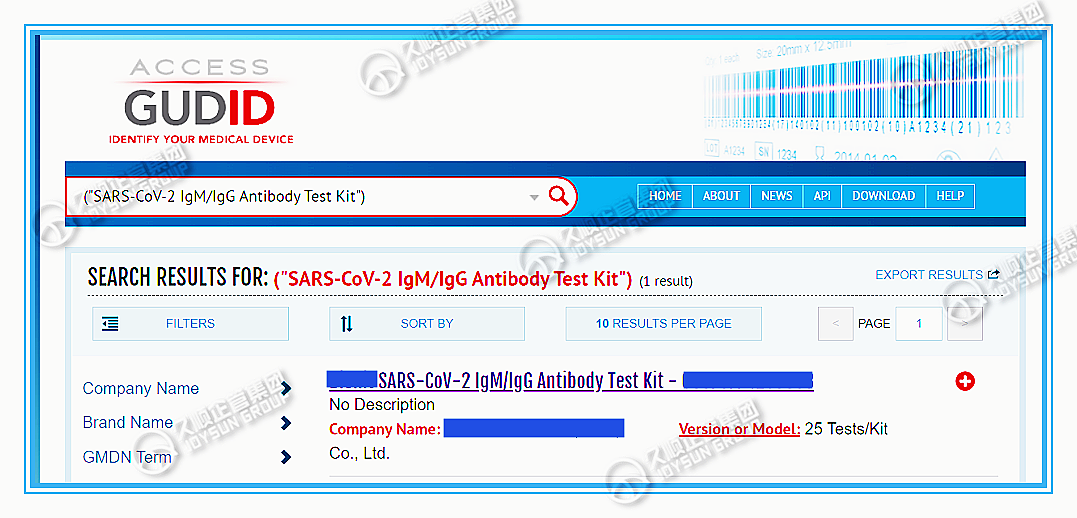
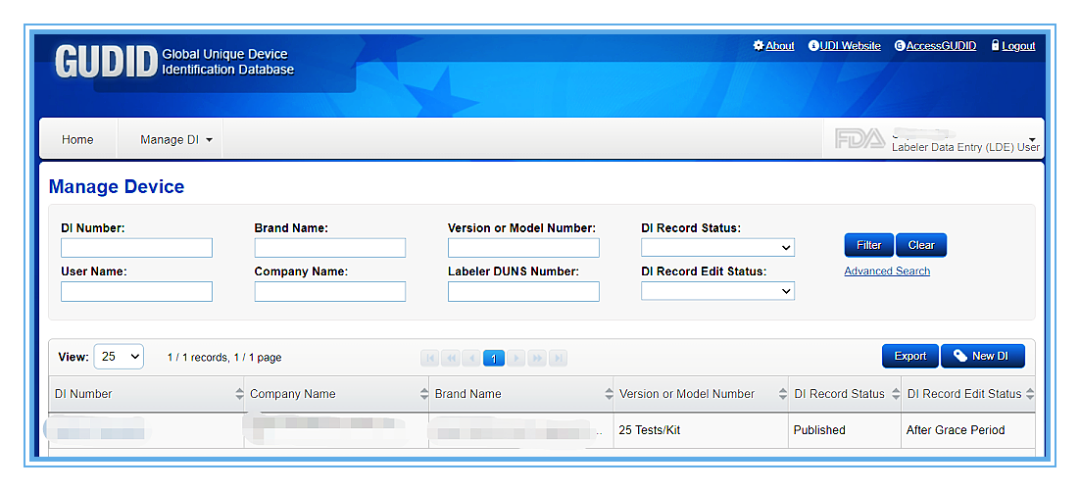
FDA已强制要求:最迟于2022年12月8日以后,所有医疗器械产品(包括Class I器械)向GUDID提交产品数据并包装上具有UDI载体。

ClassII医疗器械美国上市,制造商应注意...
大部分Class II医疗器械受到上市前通知510k或上市前批准PMA的要求约束。审查和批准510(k)申请通常需要90天,此后提交的申请将在FDA510k数据库中显示。
当制造商准备提交510(k)时,其必须向FDA提供书面证据,证明该器械等同于已获FDA批准的器械。
为证明所申请的医疗器械和市场已有医疗器械之间的等效性,应当对两种器械作对比,通常做法为测试,必要时为临床试验。
510k提交文件应考虑的有用输入,包括:文件化设计控制过程的细节、设计输入、预期用途和使用指示。
自2023年10月1日起,除豁免外,所有510(k)申请必须使用eSTAR(electronic Submission Template And Resource)在线递交。
eSTAR更注重对实验/检测过程关注,申请人应对实验/检测过程更详细描述。
ClassIII医疗器械美国上市,制造商应注意...
由于Class III医疗器械的风险水平,FDA规定:一般和特殊控制(适用于I类和II类医疗器械)不足以确保此类器械的安全性和有效性。
上市前批准PMA,需使用数据驱动的收益/风险状况对器械进行高标准的研究,以证明其安全性和有效性。
由于涉及的器械风险较高,所以相比510(k),PMA需要提供更多信息。
而上述信息的获取,一般通过实验室测试和临床试验实现,需要临床试验数据并耗费大量的时间和资源。
美国FDA的UDI创建难度不小~
不慌,有久顺!
久顺是国内少数几家、获FDA官方授权操作GUDID及UDI的美代公司,具备丰厚的FDA UDI成功实操经验,提供全系列全过程咨询服务:美国UDI-DI创建;GUDID数据库账户创建;产品UDI信息输入GUDID数据库等。
□ 同等的合理价格,久顺客户可享受更快速、更高成功率的服务;
□ 全系列流程完成最快只需15个工作日,时效与精准兼备足以傲立业界。
□ 作为资深美代公司,久顺企管已成功为多家客户操作GUDID申请、产品UDI信息上传(因受限于篇幅,以下仅展示部分实操案例):