


导 读
多年来,美容器械一直处于医疗器械法规监管的边缘地带。欧盟MDR对于常见美容器械(即附录XVI所列无预期医疗用途产品)明确实施监管审查,然而,前述产品制造商的监管义务仍显得模糊不清。对此,欧盟委员会EC已发布系列法规(含1份问答文件),澄清了非医疗用途产品需符合MDR的要求。
附录XVI描述欧盟MDR范围内6组无预期医疗用途的器械:
·隐形眼镜;
·植入物(例:乳房或轮廓植入物);
·皮肤填充剂(例:透明质酸或羟基磷灰石钙)
·塑身器械(例:抽脂);
·激光(例:用于皮肤表面修复或毛发去除);
·大脑刺激(例:经颅刺激)。
欧盟委员会可随时在附录中添加新产品组,类似产品制造商应密切关注其修改情况。
无预期医疗用途器械的过渡期影响因素
欧盟附录XVI所列器械过渡期内,制造商需考虑:临床调查需求、对产品计划开展的重大更改、是否需要与公告机构合作等因素。相关器械制造商应该充分按照过渡时间表,立即开展工作,尤其是首次遵守MDR监管框架的制造商。
根据条例Regulations(EU) 2022/2346和Regulation (EU) 2023/1194,非医疗用途产品的过渡期限要求因多种因素而异,其中包括器械风险分类因素(分类规则参阅Regulations(EU) 2022/2347和MDR附录VIII)。
无预期医疗用途器械的过渡期要求
器械分类决定合格评定路径(即必须符合法规要求才能获得产品CE标志证书)。符合性评估路径可能包括临床调查或公告机构参与等步骤,这将为过渡期增加时长。其中,附录XVI产品的过渡期限主要取决于是否必须进行临床调查。
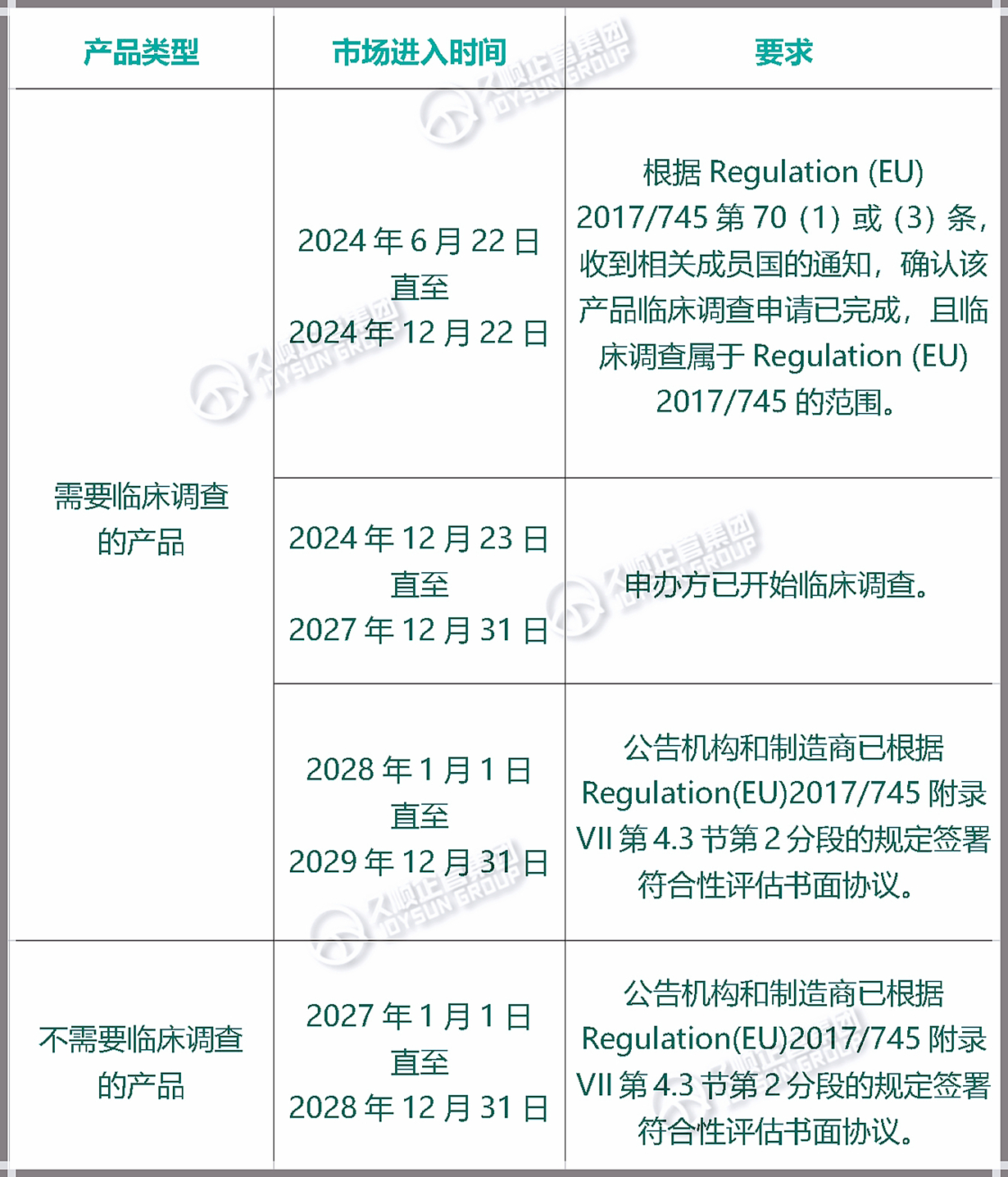
以下为:附录XVI产品过渡期时限及对应要求↓

然而,产品投放市场的时机尤为重要。欧盟监管机构希望制造商:随着过渡期临近,努力实现合规。例:如果产品需进行临床调查,并且厂商决定2024年12月至2027年12月期间将其投放市场,则临床调查必须已在进行中。
以下为:不同投放市场时间对应的符合性评估要求↓
√ 欧盟MDR\IVDR合规服务,[久顺企管]是您安心之选!
【久顺企管集团】 始创于1996年,近30年全球合规技术专家,近20年资深欧代,荷兰\英国\美国\中国均设公司,致力提供全程高效的欧盟合规服务:CE注册取证、技术文档编写、合规策略、体系辅导、上市后监督咨询等。

