


一、2023年度检查概况
2023年度,市器审中心依据《医疗器械生产质量管理规范》(以下简称《规范》)对有源类医疗器械生产企业(有源设备类,不含独立软件)共计开展了295家次现场体系核查。
二、不符合项分布情况
295家次现场体系核查共计发现不符合项1017项。从不符合项在《规范》中各章节的分布情况来看,规范的11个章节均有涉及。从各章节不符合项占比来看,前四位分别为设计开发(29.2%)、质量管理(19.4%)、采购(17.11%)和生产管理(16.4%)。
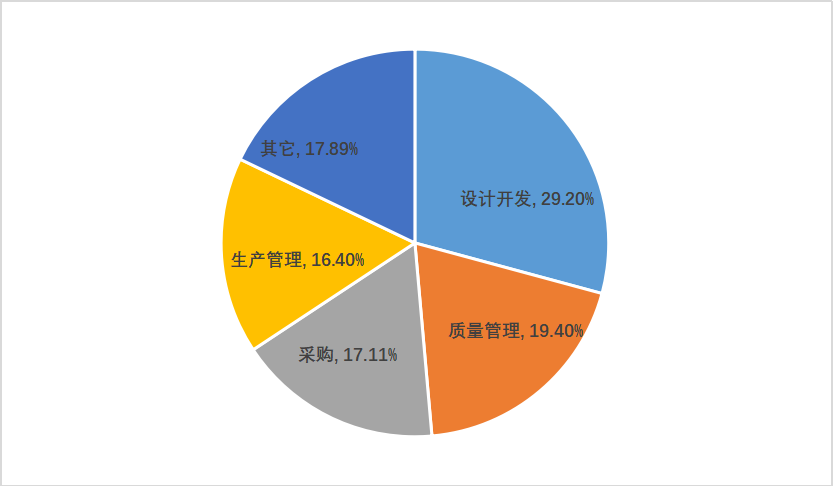
图一 高频不符合项分布情况
三、现场核查常见问题
相较于2022年,2023年度有源医疗器械现场核查发现的不符合项主要有以下变化:设计研发、采购和质量管理占比增加约2%,生产管理占比降低约2%。以下从2023年度不符合项较为集中的4个规范章节进行回顾分析。
(一) 设计开发
现场检查发现设计开发过程存在的问题,主要体现在以下几方面:
(1)部分设计开发的输出资料缺失。例如,未能提供部分物料的技术图纸、电路原理图、软件源代码等研发资料。
(2)设计验证确认不充分。部分企业的设计验证确认无法证明产品满足规定的使用要求或者预期用途。
(3)设计转移有效性不足。涉及到委托研发或研发转移时,存在着未对委托质量协议中规定的技术要求、生产工艺、质量标准等技术文件进行有效转移、接收方未能充分理解相关技术指标等问题。
(4)设计开发过程资料缺失。现场可查见设计输入和输出资料,但部分设计开发过程相关资料缺失,例如,设计开发过程中,关键部件的选型、确认、评审等过程的记录缺失。
(5)设计和开发更改未进行有效的评审、验证和确认。例如,未进行设计变更相关的风险分析并开展评审、充分识别变更的影响。
(6)创新产品的风险识别与控制不足。对于创新产品,设计开发输出的产品技术要求、采购信息、生产信息、检验作业指导书等文件中,存在着性能指标和检验方法尚无相关标准可参考的问题,企业未能进行充分有效的风险分析,将创新医疗器械产品的风险控制在可接受水平内。
(二) 采购
现场检查发现采购过程存在的问题,主要体现在以下方面:
(1)采购要求不明确。采购质量协议或采购合同中,对于主要采购部件的材质、规格型号、性能指标、质量标准、验收准则等,未明确具体要求。
(2)进口部件未实施有效的采购管理。进口部件通常存在采购合同、原材料清单、供应商资质证明文件、质量标准、检验报告及验收标准等资料不完整的问题。
(3)对于委外开发软件的供方,未作为供应商进行管理。软件委外开发协议未明确软件需求、交付形式、验收方式、维护等要求以及双方质量责任承担要求等内容。
(4)关键物料的采购记录不满足可追溯的要求。例如,关键物料未能追溯到供方批号,对于存在质量问题的物料批次,无法开展问题分析和追溯。
(三) 生产管理
现场检查发现生产管理过程存在的问题,主要体现在以下方面:
(1)生产工艺规程不合理。部分企业存在着生产过程过度依赖部件的模块化、未对产品的生产工序进行合理有效地分解细化等问题。
(2)关键工序和特殊过程的识别、验证或确认不足。关键工序和特殊过程的验证/确认报告无具体的验证方案和验证过程,未能说明重要工艺参数的来源或依据。
(3)有源无菌/有源植入等产品的生产环节控制不足。部分涉及无菌、植入等的有源产品,但生产环节控制要求不明确或充分,不满足无菌、植入等规范附录的要求。
(4)生产过程不满足可追溯的要求。未按照作业指导书的要求执行生产工艺过程并进行生产记录;生产记录不完整,未体现关键部件的序列号或批号、软件完整版本号、主要设备等信息。
(四) 质量控制
现场检查发现质量控制过程存在的问题,主要体现在以下方面:
(1)产品检验规程中的检验方法与强制性标准或产品技术要求存在差异,但企业未对所采用的方法进行科学有效的验证确认,以证明其符合强制性标准和产品技术要求。
(2)对于周期检验的项目,企业未能说明通过周期检验进行质量控制的合理性并进行风险分析。
来源:上海器审
声明:文章为转载,其版权归原作者所有。转载仅用于分享,若涉及文章版权等问题,联系我方删除!
>国内医疗器械注册,久顺是您不二之选! 近30年全球合规技术专家,专注无菌\非无菌\植入\有源\体外诊断等医疗器械领域,具备丰厚的质量体系、临床试验、产品注册咨询管理和技术服务经验,护航产品全球畅行无阻。

