


1976年5月28日美国通过联邦食品、药品和化妆品(FD&C)法案医疗器械修正案(MDA)。在MDA指导下FDA颁布法规将当时所有市售器械列为三种管制类别:I类、II类、III类。
其中,III类器械通常需要上市前批准(PMA),而如果制造商计划在美国销售预期用于人体且无需上市前批准申请(PMA)的I类、II类或III类器械,则必须向FDA申报上市前通知510(k),制造商必须在产品于州际贸易市场推出或送货前至少90天向FDA申报510(k)材料,以便FDA能够判定器械是否满足器械上市许可标准。
FDA对510(k)申报的决定基于器械与合法上市(同品种)器械是否实质等同(SE)。而在FDA颁布510(k)许可命令表明器械的实质等同前,该器械不得上市。
综上所述,对于计划开拓美国市场的制造商,寻找合适的等同产品是获得FDA上市许可的重中之重。
1.FDA对实质等同的认可标准
安全性和有效性原则是所有510(k)审查时判定实质等同的基础。FD&C法案第513(i)条规定有510(k)审查过程中判定实质等同的标准:
■ 器械与同品种器械具有相同的预期用途和技术特征,或
■ 器械与同品种器械具有相同的预期用途,但具有不同的技术特征,制造商能提供证明该器械与合法上市的同品种器械同样安全有效的信息(适当的临床数据或科学数据),这些信息不会引发与同品种器械不同的安全性和有效性问题。
“不同的技术特征”是指该器械与同品种器械相比,在材料、设计、能源或其它特性方面有着重大差异。
■ 安全性和有效性因素是FDA审查的两大部分。
首先,FDA需要发现该器械预期用途与同品种器械的“相同”,例如:器械所适用的人群或适用于治疗的疾病。
其次,器械与同品种器械作对比时,FDA还需要发现这两款器械的“技术特征相同”,或“不同的技术特征”不会引发不同的安全性和有效性问题,并且新器械与合法上市器械同样安全、有效。
在510(k)背景下,FDA一定程度上依赖于其既往判定,即同品种器械存在合理的安全性和有效性保证。FDA通过评价新器械与同品种器械之间的差异,以确定其对安全性和有效性的影响,如果新器械与同品种器械间的差异对安全性或有效性具有或可能有重大影响,那么实质等同性所需证据将随之增加。
2.查找等同器械的方法
①申请人可以在FDA医疗器械网站界面底部(如下图)进入“510(k)上市前通知数据库”。
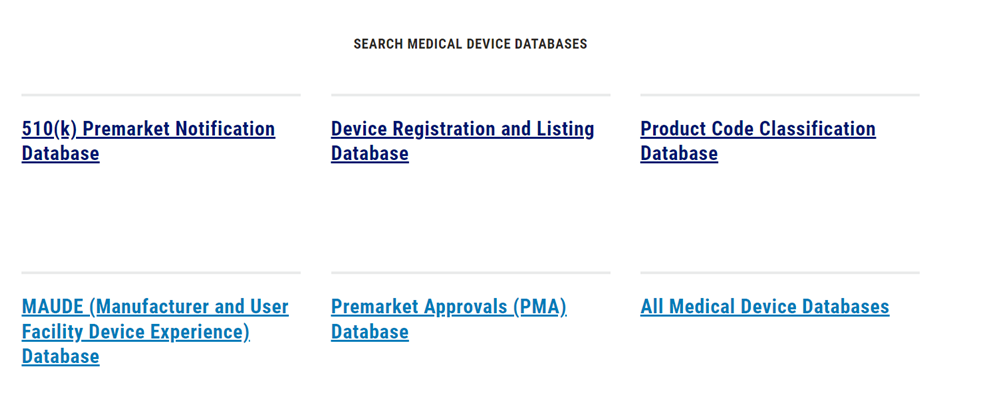
此方法尤其适用于制造商具有明确仿制对象,或明确的已知品牌的等同产品。在相应搜索框输入申请人信息或产品名称,可找到该等同产品的summary,通常1990年之后的产品都有summary,会提供详细的产品基本信息、技术特征、检测信息及明确的预期用途(具体可披露的信息由申请人确定),对制造商判断产品是否等同极具参考价值。
②申请人可以在FDA医疗器械网站界面底部进入“器械注册及列名数据库”。
此方法较适用于无预期的等同产品,但产品较成熟、名称确定,可以通过搜索同行业知名企业名称查看其列名的产品,或输入产品名称查找等同或类似产品,并获取summary,进一步确认合适的实质等同产品。
③对于较新或复杂的产品,申请人不清楚是否有等同或类似产品,那么可以通过产品的code分类,找到预期用途、适应症或技术原理较接近的产品,从而进一步查找是否有实质等同的产品。
申请人可以在FDA医疗器械网站界面底部进入产品代码分类数据库。
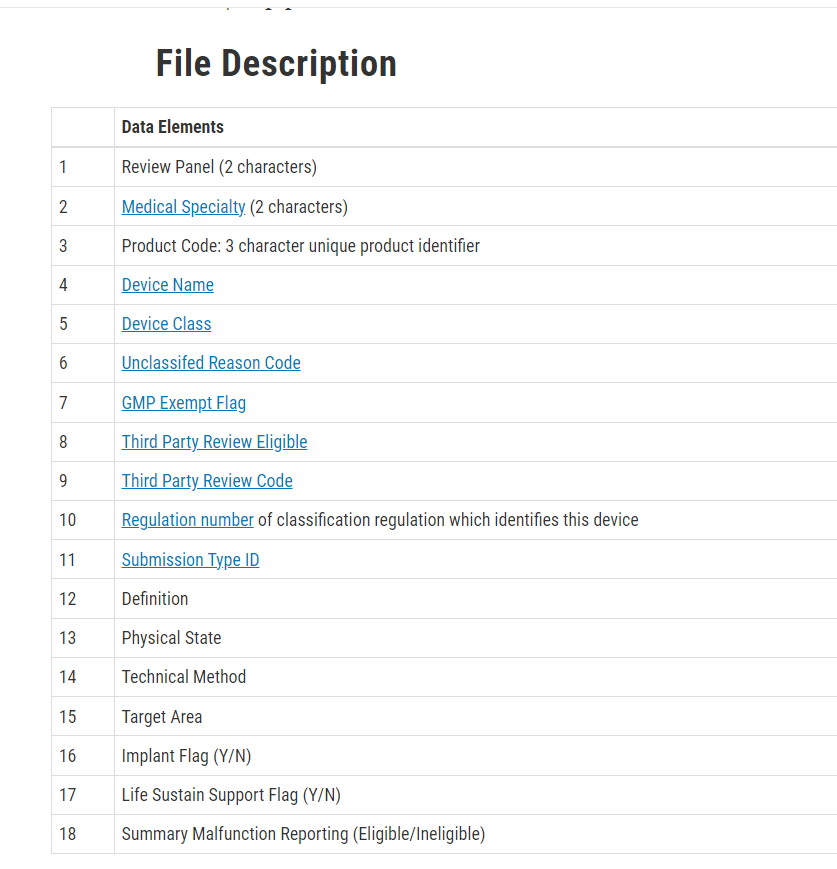
该方法需要制造商对产品code进行初步识别,依据为FDA官方的Product Code File,此文件包含以下详细信息(对制造商识别产品类别、风险及对应法规条款都较有帮助):
3.存在多个同品种器械时的选择
为判定实质等同,合法上市的器械通常被称为“同品种器械”。制造商可能发现多种同品种器械适用时(如以下示例中所描述),FDA建议在申报材料中确认1种主要同品种器械(即适应症和技术特征与受审器械最相似的器械),以便简化和促进决策流程。
多种同品种器械示例:
制造商为新的血液透析管申报510(k),该产品具有与同品种器械A相似的延长(器械在人体外的部分)设计,以及与同品种器械B相似的头端(器械在人体内的部分)设计。同品种器械A和B的预期用途与新器械相同。
# FDA 510(k)申请好帮手,非久顺莫属!#
→久顺企管集团,近30年全球合规技术专家&资深美代,成员超80%本科\硕士\博士,丰富的海外留学经验,无障碍英语口语\书面交流,一站式高效率FDA合规服务:
上市前批准(510k、特殊510k、豁免510k);创建UDI-DI\GUDID账户;QSR820体系建立维护;FDA验厂等。

