


一、临床评价的重要性
临床评价是系统的、有计划的过程,核心作用是持续地生成、收集、分析、评价医疗器械相关临床数据,目的是证明医疗器械的安全性和性能(包括预期用途中的临床疗效)。
临床评价报告(Clinical Evaluation Report,CER)必须在医疗器械整个生命周期内定期更新,并且作为技术文档的难点及核心在CE认证过程中起着举足轻重的作用。
二、MDR下临床评价的基本流程
欧盟MDR附录XIV对该要求有定义,包括:计划、数据收集、数据评价、数据生成(如有需要)、数据分析和结论、上市后阶段持续收集数据以维护和更新临床评价。
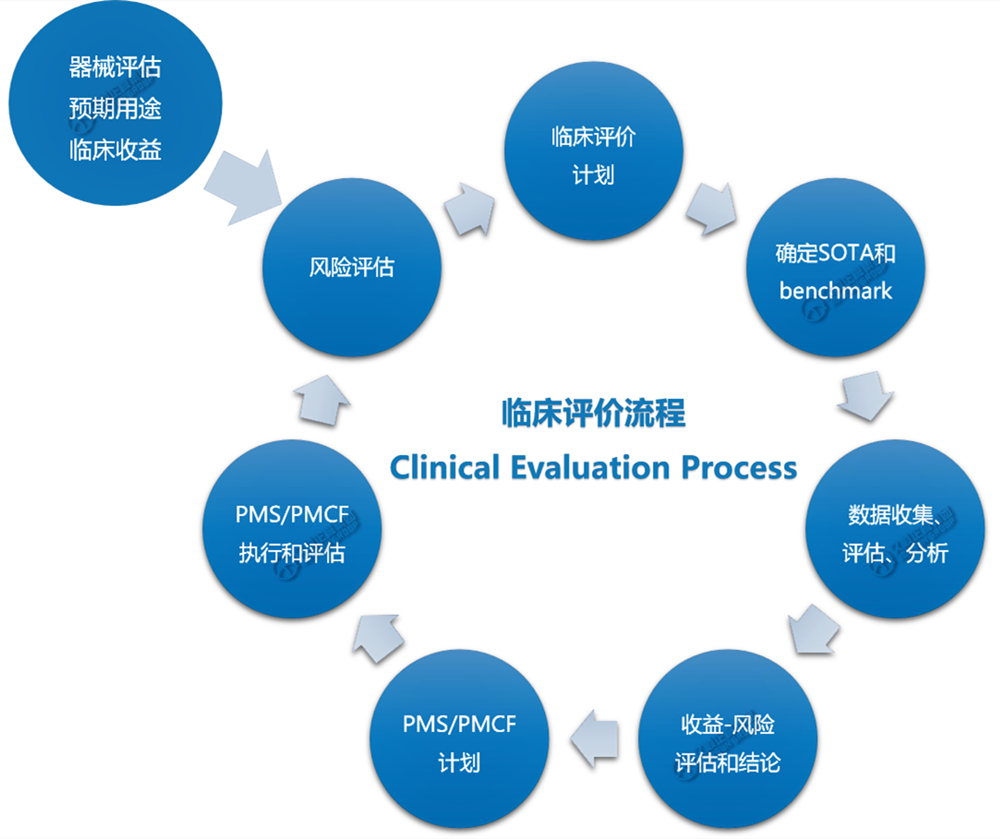
久顺总结出以下“MDR法规临床评价的基本流程”:
·建立并更新临床评价计划;
·确认临床数据、证据:通过系统的科学文献综述,确认与器械及其预期用途相关的可用临床数据及临床证据中的任何缺口;
·评价临床数据:评价所有相关临床数据,评价其对确立器械安全性和性能的适用性;
·产生临床数据:根据临床开发计划,通过适当设计的临床研究,生成新的或额外的临床数据,以解决突出问题;若遗留器械的上市后数据无法提供充分全面的临床证据,则需要生成新的临床数据;
·分析临床数据:分析所有相关的临床数据,以得出有关器械安全性和临床性能的结论,包括临床受益;
·得出临床评价报告CER;
·上市后临床跟踪PMCF。
由上述内容不难发现,MDR临床评价的核心内容或核心要求是临床数据。
三、MDR下临床数据的来源
MDR的Article 2(48)指定的临床数据来源有:
·被评价器械的临床研究;
·科学文献中报道的等同器械的临床研究或其他研究或发表于同行评议的科学文献,关于被评价器械或等同器械的其他临床经验报道;
·来自上市后监督的临床相关信息,尤其是PMCF信息。
注意事项:
1.MDR法规对临床数据来源的要求比指令更严格,使得部分指令下可被采纳的数据无法被MDR临床评价使用,例如:未经发表的其他临床经验、等同性无法被证明的器械的临床数据,不再被认为是“临床数据”。
2.MDR Article 61(3b)要求严格评价所有的临床研究结果,考虑其是否满足MDR Article 62-81和附录XV的要求,某些MDD指令下已完成的临床研究可能不符合MDR要求,此时制造商需考虑通过PMCF产生新的临床数据,而基于成熟技术或低风险的器械,可使用较低等级的临床证据表明其符合GSPR要求。
四、MDR下临床数据不足或无法提供时的应对
根据MDR 附录XIV及MEDDEV 2.7/1第四版开展临床评价时,当制造商的临床数据不足以支持所宣称的临床安全和性能时,或制造商无法提供拟评价器械临床研究数据时,经常选择与市场上已有产品作比对,证明与之等同。通过收集等同器械的临床数据用于证明拟评价器械的临床安全和性能,即等同性证明。
从Directives 93/42/EEC到Regulation 2017/745(MDR),等同性的出现次数从3增加至12,相较于MDD,MDR对等同性证明的要求更加全面和细致。
等同器械的技术文件、研究及报道资料是临床评价的重要数据来源。
→欧盟CE证书办理,快·准·好的秘诀是什么?
√答案当然是久顺企管集团!始创于1996年,荷兰\英国\美国\中国均设公司,近30年全球合规技术专家,全程高效的欧盟合规服务:CE注册取证\技术文档编写\合规策略\体系辅导\上市后监督咨询\近20年资深欧代\临床方案设计和临床试验方案等。
MDR CE成功案例(篇幅有限,仅展示部分)↓
☑辅导3H(南京斯瑞奇)取得I类灭菌创口贴的CE-MDR证书;
☑助力海翔药业取得Is类灭菌CE-MDR证书;
☑助力台衡获I类测量CE-MDR证书;
☑辅导亚美斯特(天津)获得IIa类通气类产品的CE-MDR证书;
☑辅导杭州某生物企业成功通过Is类灭菌采样拭子的MDR体系审核;
☑助力深圳客户顺利通过IIa类敷料类产品的MDR体系审核;
☑辅导某跨国医疗器械企业取得IIb类监护类产品CE-MDR证书...

