


医疗器械工艺用气,是无菌、植入性和体外诊断试剂等医疗器械生产检验过程中不可缺失的组成部分,特别是在洁净室(区)环境下使用的工艺用气,由于同医疗器械直接或间接地接触,其制备、处理和检验等过程可能影响到医疗器械的产品质量,并且洁净室(区)内使用的工艺用气若不加以控制,而直接排放在洁净室(区)内,势必对洁净室(区)环境造成污染,容易导致对医疗器械产品造成污染。
一、工艺用气的定义
医疗器械生产过程中,为满足产品不同工序的质量要求,通过一定的设备和装置制备出供医疗器械生产检验过程中使用的各种气体的总称。
常见的有压缩空气、工业氮气、工业氧气、工业氩气、工业二氧化碳等,医疗器械洁净室(区)内使用的工艺用气常以压缩空气为主。
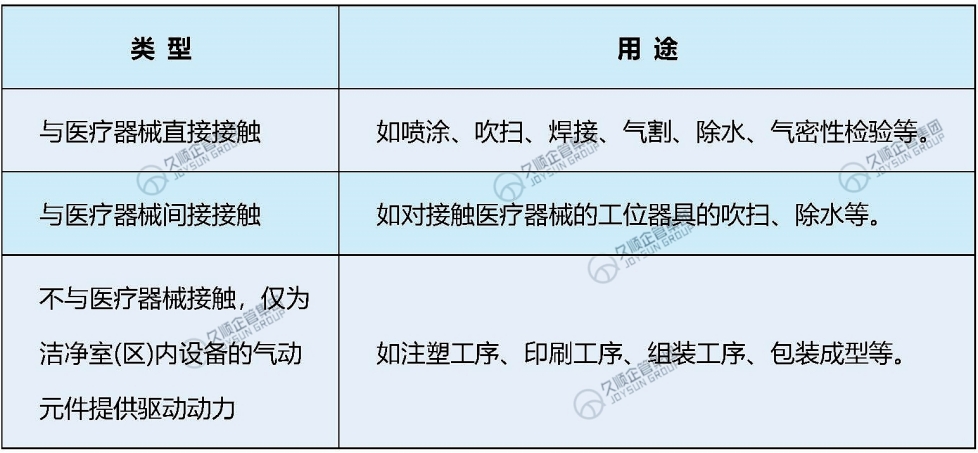
二、压缩空气的常见用途
三、压缩空气的工艺流程
一般情况下,洁净室(区)内使用的压缩空气工艺流程为:压缩空气制备→压缩空气处理(除水、除油、除菌等)→压缩空气输送→压缩空气使用。
企业应当根据压缩空气质量的不同要求及对医疗器械的影响程度,设计合理的压缩空气系统,并确定相适应的工艺流程。
四、压缩空气的主要污染源
主要包括:水(水蒸气、凝结水)、油(油雾、油蒸气)、尘埃粒子、微生物等污染物,其不利于保障产品质量,因此需要被有效地控制。
五、压缩空气的检验项目
企业应当依据产品工艺要求及压缩空气对产品质量和环境的影响程度,确定适宜的检验标准及方法。
(1)水 分
水分检测用于测量压缩空气中的微量水分,主要分为定性法和定量法,企业可根据其对产品、环境的影响程度或风险程度自行选定方法。
(2)油 分
油分检测用于测量压缩空气中的总含油量,主要分为定性法和定量法,企业可根据其对产品、环境的影响程度或风险程度自行选定方法。
(3)尘埃粒子
尘埃粒子检测用于测量压缩空气中的含尘量,为定量法,应当按照《无菌医疗器具生产管理规范》(YY0033-2000)标准执行,尘埃粒子数应当符合标准中对应的生产洁净级别要求。
(4)微生物
微生物检测用于测量压缩空气中的微生物含量(主要为浮游菌),为定量法,应当按照《无菌医疗器具生产管理规范》(YY0033-2000)标准执行,微生物数应当符合标准中对应的生产洁净级别要求。
六、压缩空气的检验周期
企业应当确定洁净室(区)内所有的压缩空气使用点,结合其对医疗器械质量影响程度和对洁净环境影响程度,定期抽取使用点进行检验,建议每季度抽取具有代表性的使用点进行1次压缩空气全项目检测,若新增压缩空气使用点则应当予以验证。
需注意:由于压缩空气为非循环输送,因此验证的使用点应当包含洁净室(区)内所有不同的使用点。
>久顺是值得信赖的体系助手! 可提供:建立并维护国内质量管理体系/ISO13485体系/美国QSR820体系/MDSAP/质量体系培训&咨询&辅导等。
√久顺企管集团:近30年全球合规技术专家,专注无菌/非无菌/植入/有源/体外诊断等医疗器械,产品注册/质量体系/欧盟CE/美国FDA/英国UKCA/欧代&美代&英代/CFS证书等具备丰厚的咨询管理和技术服务经验及能力,护航产品全球畅行。

