




警戒意味着要注意可能的危险或困难。医疗器械警戒系统旨在收集有关与医疗器械有关的上市后事故或不良事件的信息,并在适当的情况下分发或传播此类信息,以防止不良事件再次发生。
它旨在简化在使用该医疗器械的欧洲成员国之间直接、尽早且统一地实施现场安全纠正措施(FSCA)。
定义:涉及医疗器械的事故(Incidents)和现场安全纠正措施(FSCA)的通知和评估系统,称为医疗器械警戒系统。
作用:1.报告不良事件和趋势;2.执行纠正动作。
哪些属于事故(Incidents)?:市场上出售的器械的任何功能故障或性能下降,包括由于人体工程学的功能引起的使用错误,以及制造商提供的信息不足和任何不良影响。

警戒是欧盟医疗器械法规MDR第VII章中主要讨论的主题之一,其中第87-92条即由警戒系统组成。适用于警戒系统的欧盟官方指南是《MEDDEV 2.12-1rev 8 GUIDELINES ON A MEDICAL DEVICES VIGILANCE SYSTEM》。这也是EN ISO 13485:医疗器械质量管理体系 用于法规的要求 第8.2.3条(向监管机构报告)的要求。
这表明医疗器械制造商必须具有专用的系统来管理警戒活动。
如果医疗器械的特性和性能下降或使用说明书中的任何不足可能导致或已经导致死亡或患者或使用者的健康状况恶化,以及制造商出于安全原因将医疗器械撤出市场,制造商或欧盟授权代表有义务通知主管机构。

事故报告将通过非专业人士、经销商或医疗保健专业人员或从主管机构自身发送给制造商。在收到事故报告后,制造商必须首先向主管机构发送初次报告,此后制造商有义务就报告的事件进行调查。
调查可能导致制造商进行事故的风险评估,并通过现场安全通知(FSN)立即对使用者实施现场安全纠正措施(FSCA)。之后,制造商向主管机构提交最终报告,说明所采取的纠正措施,并向使用者发出安全警报。如果制造商没有对事故报告采取任何措施,他们必须在提交给主管当局的最终报告中提供正当理由。
所有报告都应以电子方式进行,上传Eudamed电子系统,向公众开放。
医疗器械警戒系统报告应根据以下标准向有关当局提交:
发生了一个事件;
该器械被认为是事故发生的诱因。
该事件导致或可能导致患者、使用者或其他人员的死亡,或者患者、使用者或其他人员的严重伤害或健康恶化。

必须同时满足以下三个条件:
-
正常使用医疗器械引起的严重事故
-
发生严重事故(死亡、严重恶化,严重公共健康威胁)
-
事件已经发生

1.使用前仪器已缺陷
2.患者自身情况导致
3.超过使用期限
4.不正常使用
5.故障但有实时警报
6.已预期会发生的副作用
7.发生几率微乎其微并未有死亡、严重恶化发生

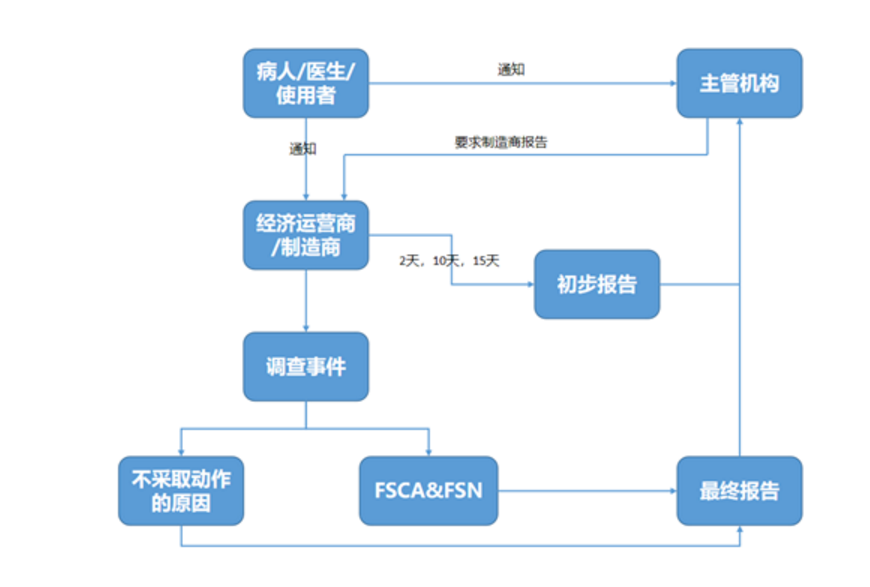
备注:严重公共健康威胁 – 2天
死亡或未预期严重恶化事件 – 10天
其他严重事件– 15天

当非严重事件发生的频率或严重程度在统计上有显著增加时或者当预期有可能对风险利益分析产生显著影响的不良副作用事件时,制造商需要通过MDR法规第92条所述的电子系统提交趋势报告。报告的是多个事件的趋势.
当权衡预期利益时,这种可能已经导致或可能造成患者、使用者或其他人员的健康或安全风险的情况是不可接受的。
这些事故应清楚地识别并记录在制造商的使用说明书中,在临床上得到充分认可,进行适当的风险评估并在技术文档中进行充分记录,并且就个人患者的利益而言在临床上是可以接受的。

制造商为减少因使用已商业化的医疗器械而造成的严重健康恶化或死亡风险而采取的措施。这些措施,无论是直接的还是间接的伤害,都必须通过现场安全通知(FSN)报告和通知使用者。
FSCA(现场安全纠正措施)是一个动作,改正或预防严重事件发生。FSCA可以是召回/器械改造/器械交换/器械销毁/患者跟进等。
FSN(现场安全通知)是一个用户通知,一份文件或表格。


特定器械的报告–导致或可能导致死亡或健康状况严重恶化的医疗器械退化或故障。




趋势报告–当非严重事件发生的频率或严重程度在统计上有显著增加时或者当预期有可能对风险利益分析产生显著影响的不良副作用事件并且权衡预期利益后这种可能已经导致或可能造成患者、使用者或其他人员的健康或安全风险的情况是不可接受的时,制造商需要通过电子系统提交趋势报告。
