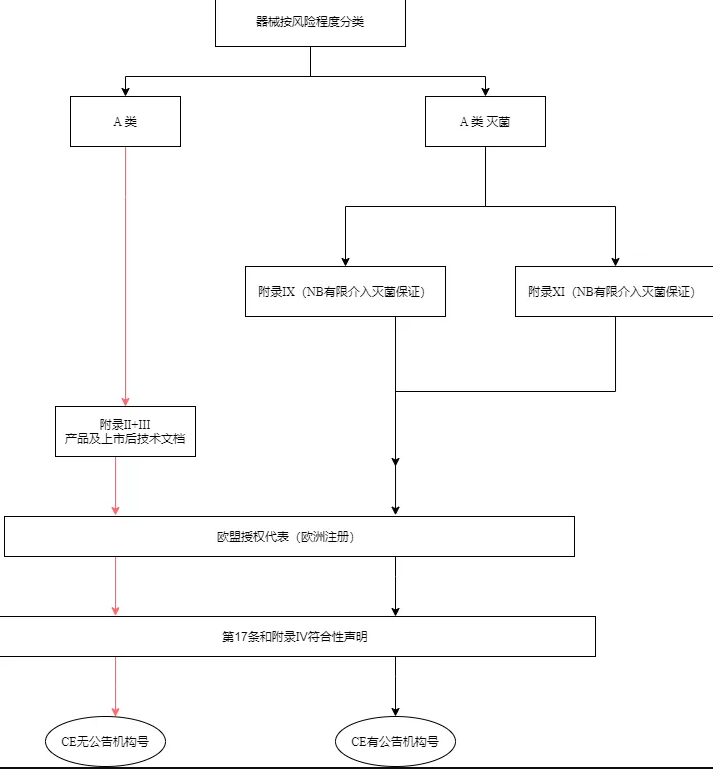
根据2017年4月5日欧盟发布的2017/746号法规,根据体外诊断医疗器械的预期用途及其固有风险,将产品分为A、B、C、D四个等级(风险程度依次升高)。若要进行体外诊断医疗器械的CE认证,首先根据产品特点及风险,确定产品的分类,产品分类确定以后还要选择该产品相应的符合性评价程序。
首先,产品在投放到市场之前,制造商应按照IVDR2017/746号法规附录IX(基于质量管理体系和技术文件的符合性评估)至XI(基于生产质量保证的合格评定)所列适用的合格评定程序对该设备进行符合性评估。各类别产品的符合性评价路径如下:
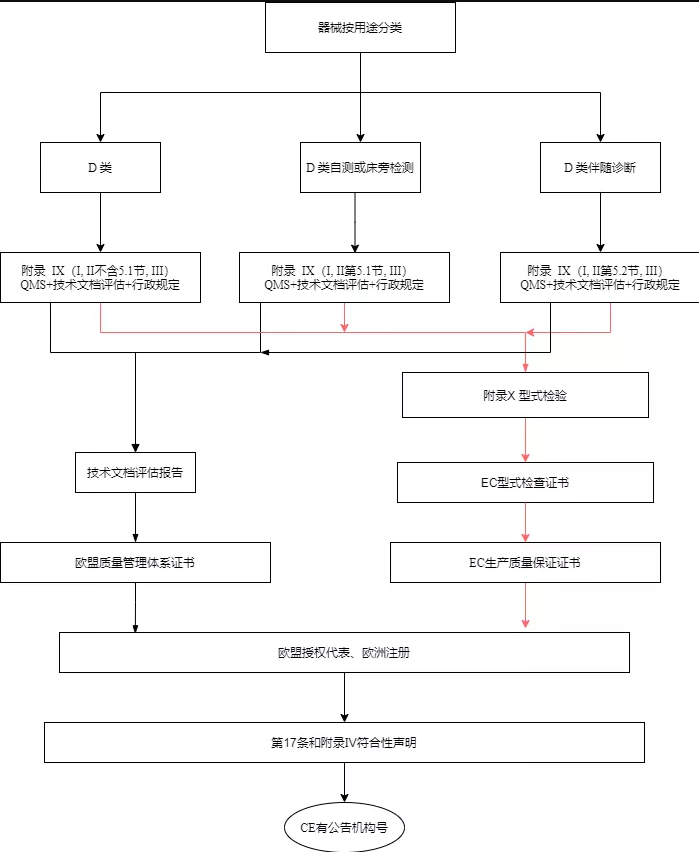
D类设备应按附录IX(基于质量管理体系和技术文件的符合性评估)的第一,第二章(第5节除外)和第三章中规定的合格评定。此外,D类自测和D类床旁检测设备的技术文档评估程序按附录IX(基于质量管理体系和技术文件的符合性评估)第5.1节中的规定进行;D类伴随诊断类设备的技术文档评估程序按照附录IX第5.2节中的规定进行。
D类设备的制造商按附录X(型式检验)以及附录XI(基于生产质量保证的合格评定)基于质量保证的合格评定中的规定进行合格性评估。对于D类伴随诊断设备,公告机构应根据附件X第3节(k)项规定的程序评定。
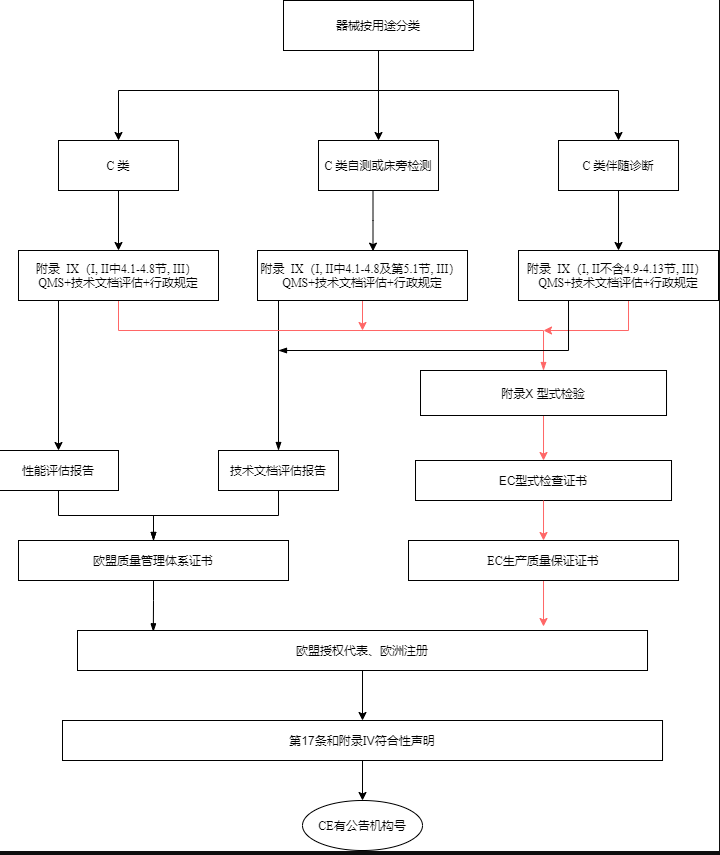
C类设备的制造商应接受附录IX(基于质量管理体系和技术文件的符合性评估)第I和III章规定的合格评定,技术文档的评定按该附录第4.4至4.8节的规定进行。C类自检和C类床旁检测设备,制造商还应遵循附录IX第5.1节中规定的技术文档评估程序;C类伴随诊断设备应采用所规定的技术文件评估程序在附录IX(基于质量管理体系和技术文件的符合性评估)的第4.1至4.8节中进行说明。
C类设备的制造商按附录X(型式检验)以及附录XI(基于生产质量保证的合格评定除第五节)中的规定进行合格性评估。
B类设备的制造商应按照附录IX(基于质量管理体系和技术文件的符合性评估)第一章和第三章的规定进行合格评定,包括该附录4.4至4.8节所规定的技术文件的评定。B类自测和B类床旁检测设备,制造商还应遵循附录IX(基于质量管理体系和技术文件的符合性评估)第5.1节中规定的技术文档评估程序。

A类设备的制造商应在草拟附录II(技术文档)和III(上市后检测技术文档)中规定的技术文件后,通过发布欧盟自我符合性声明来声明其产品的合格性。但是,如果这些设备以无菌状态投放市场,则制造商应采用附录IX(基于质量管理体系和技术文件的符合性评估)或附录XI(基于生产质量保证的合格评定)中规定的程序。建立、确保和维持无菌状态有关的方面应有公告机构的参与。