概述
欧盟新的体外诊断法规 (IVDR) 的2022年5月26日实施日期即将到来。尤其是对于那些以前被允许进行自我声明的IVD制造商,您可以在其他市场或为其他产品所做的一些工作可能适用于欧盟的IVDR合规性。
在这篇文章中,久顺小编选择对于国内IVD制造商来讲最大的两个市场欧盟和美国,从监管监督、器械分类、上市后监督、标签和临床证据的要求进行两者的比较。
FDA体外诊断法规
美国食品和药物管理局(FDA)将IVD视为医疗器械,因此它们受CFR中医疗器械相关法规的约束包括相同的上市前和上市后控制。但是,IVD也可能被视为受《公共卫生服务法》第 351 条约束的生物制品。
实验室开发测试(LDT)的进步导致对这些诊断测试的审查更加严格,目前这些诊断测试在技术上不被视为 IVD。
FDA 已发布指南草案和讨论文件,描述了某些监管要求和某些类型的LDT的执法自由裁量权。
这自然会为开发LDT的制造商和实验室创造一个灰色地带,以符合美国的IVD法规。
欧盟体外诊断法规
IVDR 的实施是欧盟监管格局的重大转变,许多制造商发现他们并没有为它所需要的一切做好准备。
如果您是IVD制造商,在我们走向IVDR申请日期时,需要注意以下一些主要变化:
-
最重要的变化之一是范围已经扩大,因此许多以前自我认证的IVD产品将需要根据IVDR进行公告机构认证。虽然有过渡期,但是IVD产品在过渡期后最终还是应获得IVDR认证。
-
新的基于风险的分类规则适用于所有IVD以代替以前的分类规则。
-
符合IVDR要求的质量管理体系。
-
将现有技术应用的标准会需满足协调标准,这势必引起一些混乱。
-
法规合规负责人(PRRC)是一项新要求,必须确定有资格担任此角色的员工并记录负责监管合规的个人。
-
经济运营商也是IVDR中的一个新术语,担任此角色的实体必须满足一定的要求,这可能是进口商、授权代表、制造商或分销商。因此需要为IVDR合规建立许多经济运营商协议和流程。
这些只是IVD制造商在您从IVDD过渡到IVDR时必须注意的一些最重要的变化。
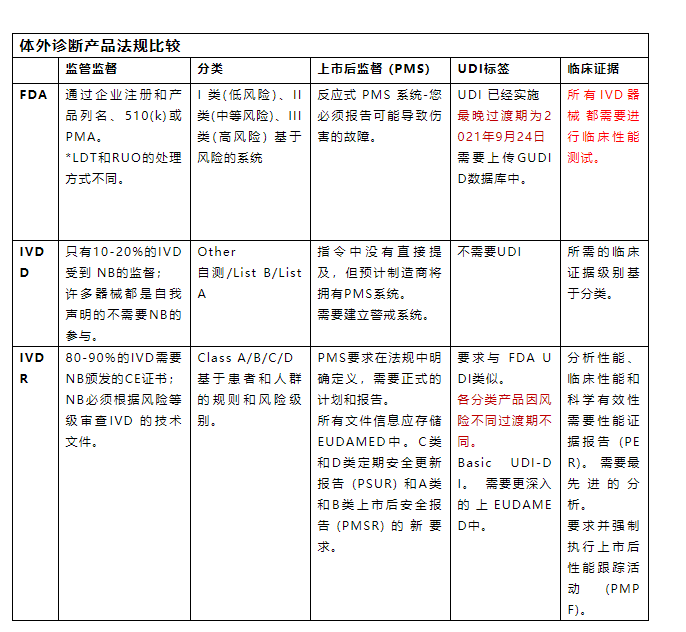
下表概述了美国和欧盟法规之间的区别。
法规的主要差异
当IVD制造商在一个市场准备提交注册时,了解不同市场不同法规框架有何不同会很有帮助。
1.监管
在美国,提交510(k)证明您的IVD基本上等同于市场上的另一种产品。FDA对其进行审查并回复一封信,说明该器械是否被视为基本等效。这是一个一次性的过程,制造商不需要证明使用最先进的技术进行持续改进,除非有设计更改需要重新提交。
在欧盟,IVD制造商现在必须向指定机构提交技术文件审查。在IVDD下,只有一小部分器械受到监管而其余器械是自我认证的。
随着IVDR的实施,大多数制造商现在必须提交给指定机构的技术文档审查。
2.分类
美国和欧盟的IVD分类不一样,没有一一对应的关系。
制造商必须遵循每个市场中基于风险的系统来对器械进行分类。
这在欧盟是新的,因为 IVDD 遵循列表系统。
IVDR的范围已扩大到包括以下所有内容:试剂产品、校准品、质控品、仪器、软件或系统以及样本容器。
3.上市后监督 (PMS)
美国IVD法规要求报告可能导致严重不良事件(死亡或重伤)的器械故障。
在欧盟对于从IVDD过渡到IVDR的制造商来说,这是一个具有挑战性的领域。
IVDR需要为PMS活动制定更加明确和严格的计划,虽然这种明确性为制造商提供了一条清晰的一致性途径,但它也给使用传统器械的制造商带来了一些挑战。
4.标签和IFU
所有市场(包括美国和欧盟)的标签和使用说明 (IFU) 要求各不相同并且必须符合相关法规。
IVDR下的更改包括在标签上包含NB编号和UDI。
5.临床证据
在美国,IVD器械的临床证据要求取决于分类但没有报告要求。
对于FDA来说,重点是制造商的验证和验证研究以支持安全性和性能。
根据IVDR,如果有正当理由,要求为您自己的器械和/或等效器械提供足够的临床证据。
对于许多拥有无法获得足够数据或不符合IVDR 要求的旧器械的制造商来说,这是一个巨大的挑战。
欧盟IVDR要求还包括上市后性能跟踪计划和报告。