概述
本期内容是MDCG 2022-8 Regulation (EU) 2017/746 - application of IVDR requirements to ‘legacy devices’ and to devices placed on the market prior to 26 May 2022 in accordance with Directive 98/79/EC的解读。
介绍
本文件提供了关于适用性的指导 “遗留器械”(legacy devices)和“旧”器械(Old devices
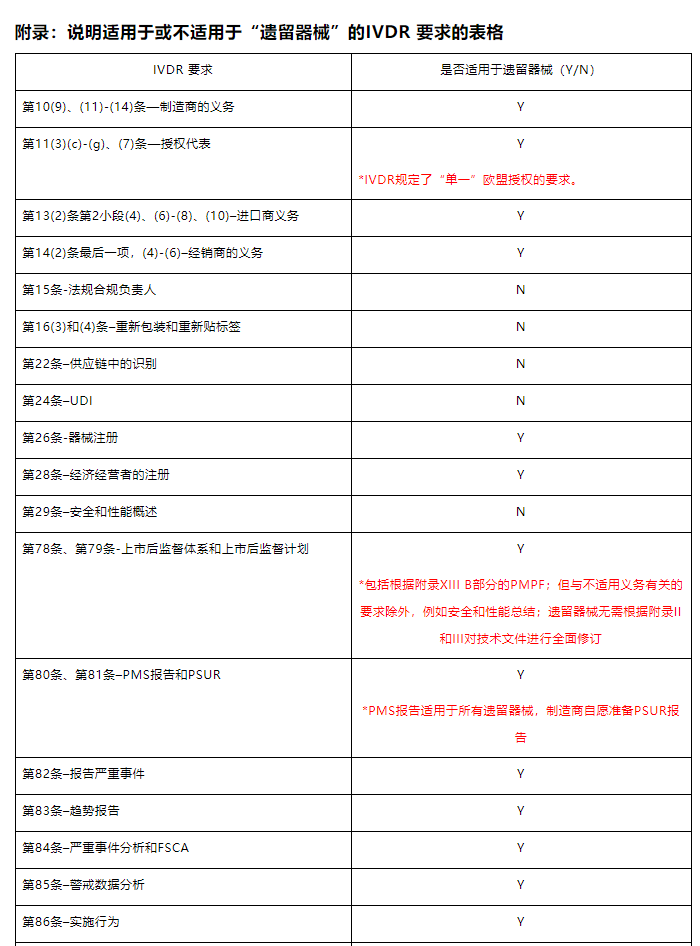
)的 IVDR 要求。附件包含一个非详尽的表格,说明适用或不适用于“遗留器械”的 IVDR 要求。帮助制造商更好的在过渡期内满足IVDR要求。
各分类产品的过渡期时间,想必各位读者已经如数家珍,小编就不在这边重申,大家可以翻看久顺过往好文。
术语
遗留器械(legacy devices):IVDR第110条第(3)款第2或第3小段所指的器械,这些器械在2022年5月26日之后投放市场或投入使用。这些器械可以是:
a)在2022年5月26日之前,由公告机构根据关于体外诊断医疗器械指令IVDD 98/79/EC颁发的有效CE证书涵盖的器械;
b)在2022年5 月26日之前根据IVDD起草的自我符合性声明并且根据 IVDR的符合性评估程序需要指定机构参与的器械。
旧器械(Old devices):指在2022年5 月26日之前根据IVDD或适用的国家规则在IVDD生效之前投放市场或投入使用且在5月26日之后仍在市场上或仍在使用的器械。
将IVDR要求应用于遗留器械
1.IVDR的上市后监督、市场监督和警戒的要求适用于遗留器械,这意味着“遗留器械”的制造商需要建立上市后监督(PMS)系统制定PMS计划执行,但更新安全和性能摘要不适用“遗留器械”。
2.作为上市后监督系统的一部分的上市后性能跟踪(PMPF)要求也适用于“遗留器械”。制造商需要“主动收集和评估器械使用的性能和相关科学数据”。
3.制造商是否需要对遗留器械完善其上市前的要求编制符合IVDR要求的技术文档?技术文档属于上市前要求是不适用于“遗留器械”。但制造商应在过渡期结束前对其产品按IVDR分类及其符合性路径获得公告机构颁发的CE证书。
4.与严重事故和现场安全纠正措施和趋势报告相关的要求和程序以及市场监督、警戒系统适用于“遗留器械”。
5.由于IVDD 98/79/EC没有规定IVDR中规定的风险等级的器械分类规则,因此,作为最低要求,PMS 报告应适用于所有遗留器械,除非属于C或D类的“遗留器械”的制造商自愿准备PSUR报告。
6.关于根据IVDD由公告机构颁发的证书所涵盖的遗留器械(List A/B/Self-test),在其监督活动过程中,公告机构应考虑到因过渡条款而适用于制造商的新要求。在审查适用要求作为其“适当监督”的一部分时,在指定机构的参与方面需要有灵活性。
7.经济运营商的义务和UDI/器械注册的要求同样适用于遗留器械,其他的IVDR要求原则上不应适用于“遗留器械”。
将IVDR要求应用于2022年5月26日之前投放市场的器械(旧器械)
IVDR要求原则上不适用于旧器械。
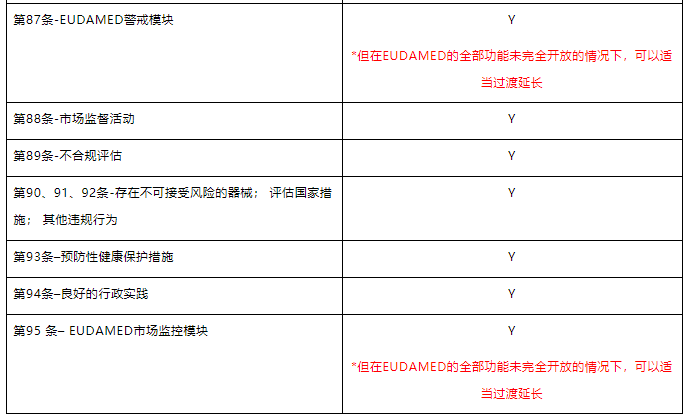
但是,IVDR中第88至95条规定了主管当局在市场监督活动方面的权利和义务适用于2022年5月26日之后的旧器械,这允许主管当局检查这些器械是否符合投放市场时适用的规则并对不合规或不安全的器械采取适当措施。
此外,2022年5月26日之后发生的与旧器械有关的严重事件和现场安全纠正措施的报告和分析应按照 IVDR进行。